

最後更新於 2023 年 7 月 11 日
食品添加物在台灣共分為17類,而且需要得到許可才可販售及使用,屬正面表列之性質,而不同的國家會有不同的分類及使用規範,故需要了解販售國對食品添加物的規定及限制。為使台灣食品添加物之增列及修訂可明確化,衛生福利部食品藥物管理署特定訂【食品添加物使用範圍及限量暨規格標準增修訂申請作業指引】以供遵循,目前先發出草案供業者參考及討論,欲裨益指引可更加完善。
食品添加物使用範圍及限量暨規格標準增修訂申請作業指引(草案)
壹、指引說明
一、目的
衛生福利部依據食品安全衛生管理法(以下簡稱食安法)之規定,訂有「食品添加物使用範圍及限量暨規格標準」,規範食品添加物之品名、規格及其使用範圍、限量標準,該標準係正面表列,表列上之食品類別得依限量規定合法添加使用,非表列上之食品類別與未准許之食品使用範圍,則不得使用。業者若有申請增列或修正食品添加物使用標準之需求,應依申請項目,備齊「食品添加物使用範圍及限量暨規格標準增修訂申請表」列載相應資料,如涉及使用範圍與限量之增列或修正,需提供國人膳食風險評估資料,送予食品藥物管理署(以下簡稱食藥署)辦理審查與行政作業程序。所送資料經確認完整無誤後,依食安法之規定送交食品衛生安全與營養諮議會審查,若獲通過,將辦理草案預告徵詢各界意見,俟完成各項程序後正式公告實施。
本指引目的為業者在準備提出食品添加物之增列或修訂申請案時,提供申請文件中所需要必備文件(食品添加物使用範圍及限量暨規格標準增修訂申請表、應檢具資料等),使業者可依循。
二、範圍
本指引中「申請者」是指有提交申請之法人、機構或團體,例如:食品業者、食品公協會及顧問公司等。
貳、申請案格式
一、介紹
申請案內容應敘述作為評估食品添加物安全性的依據,內容需包含足以支持食品添加物標準增修訂內容之相關研究描述與數據,必備資料詳見附件一「食品添加物使用範圍及限量暨規格標準增修訂申請表(以下簡稱申請表)」。
食品添加物增修訂申請案需符合申請表中所有可蒐集到之資料,以便對標準增修訂進行審核評估,並附上紙本資料。若申請文件具保密性,該申請案還需檢附必須保密之文件清單以及必須機密處理之原因與可驗證依據。
不具保密性之資料需有完整電子檔案,申請時檢附CD/DVD/USB,於後續通知時需有兩個版本,一是完整版本,另一為保密性版本。
二、申請案注意事項
編寫申請案內容時需考量之注意事項,請參閱下述說明。
(一) 格式
應檢具資料宜使用Microsoft Word 或兼容的文字處理軟體編寫。標題與內文之中英文分別為14 號字之標楷體與Times New Roman,紙張大小為A4,頁邊界為2.5 公分,單行間距,每個標題下段落的第一行縮排2 字元,段落之間無間距,左右對齊,不同研究描述之間應增加額外行距。英文單字(包含半形括號)中間需空格,中文單字與英文單字中間不留空格。
(二) 計量單位
應使用國際公制單位(Système International d’Unités, SI units),包含以毫克/公斤(mg/kg)代替百萬分之一(ppm);放射性以貝克(Bq)代替居里(Ci)。壓力單位為例外,毫米汞柱(mmHg)應在括號中標示相同壓力的千帕(kPa)。
當以毫克/公斤(mg/kg)表示濃度時,應避免與單位體重攝取量混淆(mg/kg bw)。數字與單位之間沒有連字符號,但需有空格,例如:應寫為0.5 kg 大鼠,而非0.5kg 大鼠或0.5-kg 大鼠。單位與斜線(/)間不應有任何單詞。例如:應寫為果膠攝入劑量為每公斤體重3 微克(3 μg/kgbw),而非每公斤體重3 微克果膠(3 μg pectin/kg bw)。
(三) 劑量表示方式
在表示實驗劑量時,應將換算後之劑量描述於括號中,例如:以管餵方式餵食實驗動物x mg/kg 之抗壞血酸(等於或相當於實驗動物雄性攝入a mg/kg bw-day 與實驗動物雌性攝入b mg/kg bw-day)。
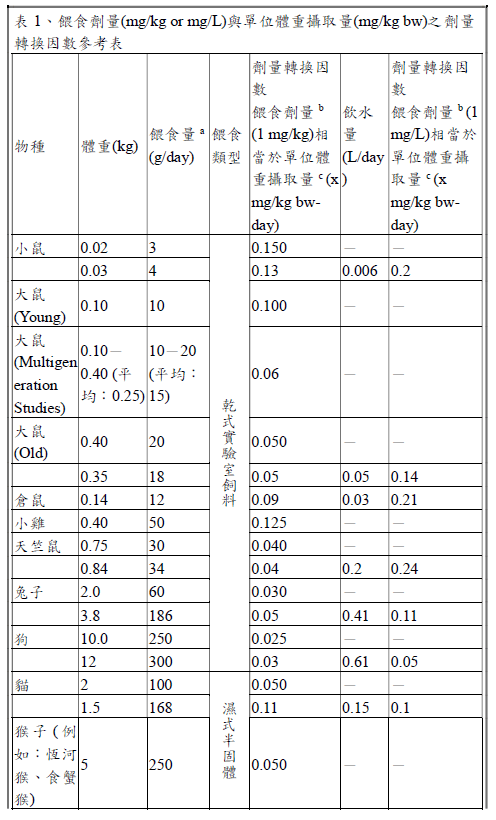
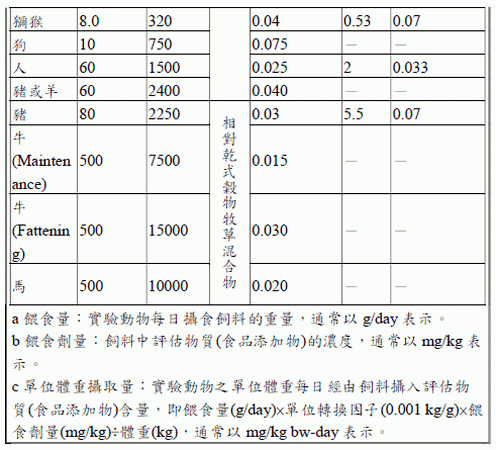
當安全性試驗研究有提供實驗動物之攝食量(即每日攝食多少飼料,g/day 或L/day)、餵食劑量(即飼料中食品添加物之濃度,mg/kg 或mg/L)及實驗動物體重(kg)等數據,可直接推算實驗動物單位體重攝取量(mg/kg bwday),使用「等於(Equal to)」來描述單位體重攝取量(JECFA, 2016)。
舉例說明,實驗動物為0.025 kg 小鼠,以管餵方式餵食每日2 g (2 g/day)含500 mg/kg 抗壞血酸之飼料(等於攝入40 mg/kg bw-day);計算如下:單位體重攝取量=攝食量(2 g/day)×單位轉換(0.001 kg/g)×餵食劑量(500mg/kg)÷動物體重(0.025 kg)= 40 mg/kg bw-day。
若安全性試驗研究無提供實驗動物攝食量或體重,僅提供餵食劑量(mg/kg 或mg/L),則需以劑量轉換因數推估單位體重攝取量(mg/kg bw-day),可參考以下參考表,則使用「相當於(Equivalent to)」來描述單位體重攝取量(JECFA, 2016)。
舉例說明,實驗動物為小鼠,以管餵方式餵食含1000 mg/kg 抗壞血酸之飼料(相當於攝入150 mg/kg bw-day);計算如下:根據參考表,小鼠體重假設為0.02 kg,餵食量為3 g/day,單位體重攝取量相當於餵食劑量(1000 mg/kg)×劑量轉換因數(0.150),即相當於150 mg/kg bw-day。
如有必要,轉換劑量時最多可增加兩位有效數字,以避免在計算時因四捨五入造成不確定性。可在研究描述時進行劑量轉換,原始劑量可表示為mg/kg、mg/L、攝食百分比等,但不能表示為ppm,如果有ppm必須更改為mg/kg 或mg/L。
(四) 參考文獻
1.內文中參考文獻可能有一位(Brown, 1999)、兩位(Brown & Jones, 1999)、或以第一作者加等人來代表三位或更多作者(Brown et al., 2000)。請注意使用&而不是and 一詞,並在年份之前使用逗號。
2.若同一作者在同一年發表了不只一篇參考文獻,則應使用a、b 等來區分參考文獻(Jones & Brown, 1999a, b; Smith, 2000b)。該規則也適用於 “et al.”參考文獻,即使每個參考文獻中的其他作者都不相同(Brown et al., 1999a,b)。
3.同一姓氏的不同作者在同一年發表論文,則應使用名字之首字母區分參考文獻(Li R et al., 2000; Li Y et al., 2000)
4.參考文獻內文中按時間順序遞增引用(但同一作者的所有參考文獻一起列出),並對同一年出版文獻時按字母順序引用(Brown, 1988,2003; Brown & Smith, 1989; Smith & Brown, 1989, 1991; Brown et al.,1991; Jones, 1999a,b)。
(五) 專有名詞
當內文出現專有名詞,應在首次出現專有名詞後括號敘明原文全名與縮寫,之後可僅使用縮寫,例如:
1.某食品添加物於國際食品法典委員會(Codex AlimentariusCommission, Codex)之限量標準。
2.以彗星試驗(Comet Assay)評估某食品添加物對生殖細胞DNA(Deoxyribonucleic Acid)結構之影響。
3.人類研究(ヒトにおける知見),包含:流行病學調查、臨床試驗、案例研究觀察、職業暴露。
(六) 其他注意事項
申請案文件力求簡潔,只需提供與增修訂申請案件評估所需要之內容,應避免置入過多不相關之研究或資料,例如:安全性試驗資料不應放入食品添加物對健康之效益等。
參、申請案需準備內容
食品添加物使用範圍及限量暨規格標準增修訂申請,可參考申請表(附件一)撰寫,於符合申請表之項目勾選註記,此外需附上「應檢具資料」之摘要與完整資料,包含該食品添加物的規格標準、食品添加物製造方法、食品添加物規格與檢驗方法、食品中食品添加物之檢驗方法、世界各國法規標準及准用情形、技術性文件、安全性試驗資料、其他國家或國際組織之風險評估報告、依據國人膳食習慣之風險評估、在人類上之研究與發現、其他必要性資料、無法檢具上述資料之說明等,及應檢具資料中所引用的參考文獻。
故一份完整食品添加物標準增修訂申請案必須包括:食品添加物使用範圍及限量暨規格標準增修訂申請表、應檢具資料之摘要、應檢具資料、參考文獻。
一、食品添加物使用範圍及限量暨規格標準增修訂申請表
申請業者必須填寫並提交申請表(附件一),並勾選申請表中各項目以檢核所檢具之檔案是否完整。申請表內容分為:申請業者相關資料、申請項目及應檢具資料等三部分。
(一) 申請業者相關資料
申請者應於表格內填入詳細申請者、聯絡人、電話、地址及申請日期。
(二) 申請項目
申請項目分為「1. 增列食品添加物新品項」與「2. 修正目前本國准用食品添加物之規範(其中細分為2A.修正現行使用範圍及用量標準、2B.修正現行規格標準)」,於申請表中擇一勾選。
申請項目為「1. 增列食品添加物新品項」,需注意以下事項:
1.食品添加物品名:需填寫中英文名稱。
2.申請之食品添加物功能及分類:需依我國「食品添加物使用範圍及限量暨規格標準」18 類用途填寫。
3.申請之使用食品範圍及限量:參考食藥署現行食品使用範圍及限量內容之格式撰寫。
4.申請原因:需填寫欲申請增列之食品添加物於食品中之功能與需增列食品添加物新品項之原因等。
申請項目為「2. 修正目前本國准用食品添加物之規範」,需注意以下事項:
1.食品添加物分類及品名:需填寫中英文名稱。
2.申請修正項目:若修正項目為修改現行食品添加物使用範圍及用量標準,需說明修改之項目,可參考食藥署現行使用食品範圍及限量標準之格式撰寫。
3.申請原因:需填寫欲申請修正食品添加物之現行使用範圍及用量標準之原因等。
(三) 應檢具資料
依照申請項目之類別確認蒐集應具備之資料附件項目,並勾選所蒐集到之資料,詳細需蒐集與準備之內容詳見以下說明。
二、應檢具資料之摘要
應簡單說明所附資料之重點,如世界各國法規標準及准用情形、技術性文件內容、安全性試驗資料、及其他國家或國際組織之風險評估報告之簡要說明等。
三、應檢具資料之項目說明
應檢具資料中必須包括規格標準、食品添加物製造方法、食品添加物規格檢驗方法、食品中食品添加物之檢驗方法、世界各國法規標準及准用情形、技術性文件、安全性試驗資料、其他國家或國際組織之風險評估報告、依據國人膳食習慣之風險評估、在人類上之研究與發現、其他必要性資料及無法檢具上述資料之說明。若申請增列食品添加物新品項,需盡可能提供完整之檢具資料,應檢具資料項目說明如下。
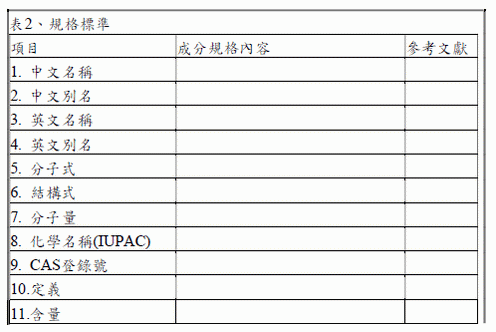
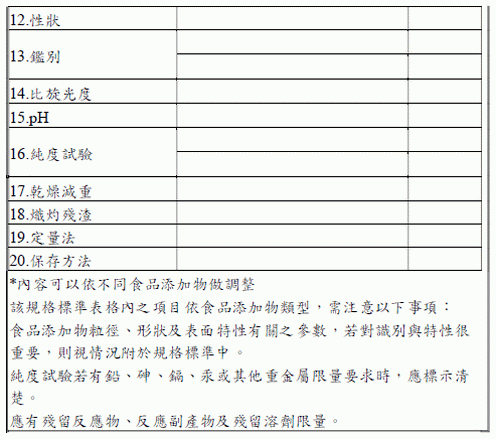
(一) 規格標準
為確保食品添加物的安全性與品質,需提供世界各國規格標準、規格檢驗方法之法規依據,若有特別之使用限制、備註、或說明亦須列於附錄中,例如:IUPAC 命名、英文別名、分子式、分子量、純度、物化特性、外觀性狀、重金屬等不純物之規範等項目,可優先參考Codex 與JECFA,倘Codex、JECFA 未有相應之規格標準,則可參考歐盟、美國或日本等先進國家所訂食品添加物規範之內容,規格標準亦需附加參考文獻,若沒有現有規格標準時,需新建立該食品添加物的規格標準。屬於申請項目2.A 者得免附。規格標準可參閱以下範例。
(二) 食品添加物製造方法
因食品添加物製造過程,可能會有相關化學物質之殘留,但非該食品添加物之成分,例如:起始物質殘留、副產物、製造食品添加物之反應物及添加物之分解產物等,天然來源之添加物則可能含有已知有毒物質。
由於不同的製造過程可能導致不同類型或含量之雜質,因此應簡明地描述製造方法,說明如何於製造過程中控制、減少或去除雜質、副產物或毒性物質。可使用流程圖等簡單地記載製造流程,危害因素的去除流程也應一併記錄。
另可記錄食品添加物是何時、於何國家被開發、於何國家作為食品添加物,及描述從申請到被使用為止的經過。若屬於申請項目2.A 者得免附。
(三) 食品添加物規格檢驗方法
食品添加物規格檢驗方法透過物理、化學、或生物學方法,檢測其物理、化學性質、鑑別試驗、有效成分含量、雜質、或污染物限量。若已設定檢測方法後,應確定檢驗中使用參考標準品之規格,如果有公開之食品添加物規格檢驗方法則可適當引用。
建立方法時應特別注意準確性、可重複性及特異性。即使該食品添加物的特異性低,若可透過混合其他適當的物質,測試出相對含量,則建議採用來確保測量的可重複性。
如果要確定的成分不止一個,則應按重要性依序進行描述。若屬於申請項目2.A 者得免附。
(四) 食品中食品添加物之檢驗方法
針對使用該食品添加物之食品,提供食品中食品添加物之檢驗方法。至少應建立一種以上之食品中食品添加物定量與定性分析方法,例如:針對該食品添加物食品之特定化學分析方法,檢驗方法必需可以區別食品中其他相同添加目的之食品添加物。
如果無資料,請於(十二)無法檢具上述資料之說明補充說明。屬於申請項目2.B 者得免附。
(五) 世界各國法規標準及准用情形
描述該食品添加物於世界各國或國際組織的准用情形,包括:法規准用的食品名稱、規格、使用範圍及限量標準,業者除了列出申請食品添加物之食品項目與使用限量外,還需附加該食品添加物於其他食品申請的使用限量。
可參考制訂相關食品法規較為完善之國家或國際組織,例如:Codex、歐盟、美國、紐西蘭、澳洲、日本及韓國等,如下說明:
1.Codex:在GSFA 中,有規定每種食品的使用限量等,可於GSFA online 中查詢簡易資訊。因維生素與礦物質在Codex 中並未作為食品添加物管理,所以在GSFA 中沒有規定使用標準,可參考訂有規範之其他個別商品標準(Commodity Standard)。
https://www.fao.org/gsfaonline/additives/results.html?lang=en
2.歐盟:Regulation (EC) No 1333/2008 的附表II 中,規定每種食品的使用限量等,且附表II 定期修訂。
https://eur-lex.europa.eu/homepage.html
3.美國:聯邦規章典集(Code of Federal Regulations, CFR)的主題21(21CFR)中規定每種食品的使用限量。除此之外,GRAS Notice Inventory 紀錄有在食品GRAS 物質的項目。在美國,維生素與礦物質是依據膳食補充劑健康與教育法(Dietary Supplement HealthEducation Act; DSHEA)管理。
4.澳洲與紐西蘭:於食品標準法典(Food Standards Code)標準1.3.1 的目錄(Schedule) I-IV 中,規定食品添加物使用限量。目錄I 紀錄除食用色素外之食品添加物使用限量;目錄II 中記錄未規定使用限量之食品添加物;目錄III 紀錄未規定使用限量之食用色素;目錄IV規定了每個食品的使用限量等。
https://www.legislation.gov.au/
5.日本:可參考厚生勞動省之既存食品添加物名簿上訂定之食品添加物之規格標準、使用範圍及限量標準。
https://www.mhlw.go.jp/index.html
6.韓國:MFDS 依據Food Additives Code 訂定食品添加物之規格標準、使用範圍及限量標準。
http://www.mfds.go.kr/eng/index.do
(六) 技術性文件
應包括理化特性、使用用途、其他類似之食品添加物效果比較、在食品中之穩定性、預期用途與使用量之研究,例如:抗氧化劑測試,應根據其使用量與經過時間來闡明對食品的抗氧化作用;防腐劑測試,應闡明食品添加物在改善食品防腐功能的有效性;欲增修訂之食品添加物可與現行法規已准用之食品添加物進行有效性的比較。
應檢查食品添加物在食品中的穩定性,若不穩定則應檢測主要分解產物的種類與數量,還應檢測食品添加物對食品主要營養成分的影響,為了證實食品添加物具有預期效果,以確認食品添加物的利用目的,需要提供相關實驗設計的試驗依據與階段性地添加於食品中食品添加物的量。
此外亦可記錄每個階段的效果,與不使用食品添加物的情況互作比較,進行顯著性評估等適當的統計處理確認有效性,這些資料不僅用於有效性的實證,也期望用於計算出最小有效利用量。若屬於申請項目2.B 者得免附。
盡量不要用「在國外廣泛使用」等模糊字眼敘述,最好附上最低限度與已經可使用的同樣用途之食品添加物相比之技術性文件資料,並說明其差異或優點、添加用途、作用機制、反應機制及文獻資料等具體說明。若屬於申請項目2.B 者得免附。技術性文件應具備以下項目:
1.食品中穩定性試驗:應提供證明食品添加物安定性之數據,特別是若食品添加物對環境敏感條件(水分、空氣及溫度)或穩定性有限,穩定性試驗應在食品添加物預期使用時間內之預期使用條件下進行。
2.預期用途與使用量之研究:若使用此項食品添加物可以達成多種技術效果。例如:某食品添加物可同時做為抗氧化劑、調味劑、乳化劑或增稠劑,為達到物理或技術功能,刻意添加到食品中之食品添加物添加量不得超過在食品中達到預期技術效果之合理量,有關食品添加物的預期用途與使用量研究,包括:
(1) 何種類型之食品將使用此食品添加物,申請者具有提供數據之責任,證明所有申請之使用狀況都是安全的。如果申請者欲明確規定使用目的,需在申請案中說明,例如:本品可使用於糖蜜及糖飴;用量以SO2 殘留量計為0.30 g/kg 以下。
(2) 預計使用食品添加物之各種食品中的使用量。
(3) 明確說明食品添加物於食品中之預期技術效果,如果添加劑之技術效果與粒徑有關,其如何影響功能(溶解度、黏度、穩定性及抗氧化力等)。
(4) 食品中添加物之降解或宿命,包括在儲存或加熱過程中將形成降解產物之量。
(5) 其他推薦、建議及使用說明,包括樣品與標籤。
(七) 安全性試驗資料
需盡可能提供完整之安全性試驗資料,以JECFA、EFSA 及USFDA等國家或國際組織評估之資料優先。
申請項目若為國際間皆尚未准用之品項,應檢具完整之安全性試驗資料,若欲申請食品添加物可能有生殖毒性、致畸、致癌性及基因毒性等應特別標註。
亦須提供食品中食品添加物之降解或宿命(Fate),包括成分鑑別或在儲存與加熱過程中將形成降解產物之含量。若屬申請項目2.B 者得免附。
安全性試驗資料根據研究類型可分為急性毒性試驗、亞慢性毒性試驗、慢性毒性試驗、致癌性試驗、生殖毒性試驗、致畸試驗、致癌性試驗、基因毒性試驗、過敏性試驗、神經毒性試驗及代謝與藥物動力學研究等,如下說明:
1.急性毒性試驗:急性毒性的結果最好用LD50 表示。
2.亞慢性毒性試驗:為28 日與90 日重複給藥的毒性試驗結果,在實施90 日重複給藥毒性試驗的情況下,可以省略同一動物種類的28日重複給藥毒性試驗。
3.慢性毒性試驗:1 年以上重複給藥毒性試驗與1 年重複給藥毒性/致癌性合併試驗的毒性試驗結果。
4.生殖毒性試驗:在繁殖試驗(多代生殖毒性試驗)中,紀錄雌雄動物的生殖功能、發情週期、交配行為、受孕、分娩、哺乳、幼年動物的成長及行為等相關資訊。
5.致畸試驗:檢查試驗動物的一般狀態、體重、攝餌量、飲水量、親代動物及胎兒的剖檢結果等。
6.致癌性試驗:檢查長期間攝取食品添加物是否會引起癌症之試驗,觀察試驗動物是否產生癌症,與產生癌症反應之臟器,可包含肉眼與病理組織學檢查。
7.基因毒性試驗:以遺傳毒性整體相關的試驗結果為基礎進行評估。
8.過敏性試驗:需要實施以延遲過敏為指標的致敏性試驗,但可以利用天竺鼠皮膚過敏性試驗(Guinea Pig Maximisation Test, GPMT)或小鼠局部淋巴結分析(Local Lymph Node Assay, LLNA)。
9.神經毒性試驗:當暴露化學物質或其代謝產物而直接影響神經系統運作,或透過對其他系統間接作用於神經系統所引發之不良反應,即為神經毒性。
10.代謝與藥物動力學研究:觀察試驗動物經攝入欲評估食品添加物後,從而獲得關於其吸收、分佈、代謝及排泄等體內動力資訊,以估計人體內有害作用的表現等。
另可透過蒐集國際間各國家或國際組織之綜合安全性評估報告或資料庫取得毒性試驗資料,若無資料亦可蒐集國際上公開發表之期刊文獻或自行研究之試驗資料,並依不同毒性試驗彙整資料,常引用之國家或國際組織如下:
1.聯合國糧農組織與世界衛生組織食品添加物聯合專家委員會(JointFAO/WHO Expert Committee on Food Additives, JECFA)
https://apps.who.int/food-additives-contaminantsjecfa-database/
2.歐洲食品安全局(European Food Safety Authority, EFSA)
3.美國環保署整合性風險資料系統(Integrated Risk Information System,IRIS)
業者可參考上述國家或國際組織評估資料,依據不同毒性試驗整理內容,需提交之內容可參考以下範例。
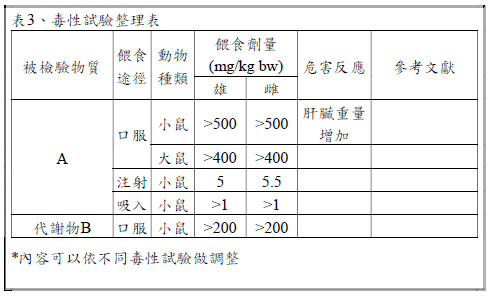
(八) 其他國家或國際組織之風險評估報告
需提供國際間風險評估報告中所計算食品添加物之最大允許濃度、添加濃度或實際檢出濃度,以及含該食品添加物之食物品項攝食量數據,彙整成風險評估報告,風險評估報告內容係整合危害鑑定、危害特徵描述、暴露評估及風險特徵描述之結果(可參閱本指引p.28 至p.38 所述內容),如該食品添加物無危害特徵描述相關數值(例如:ADI 或MTDI),可提供暴露評估報告。以JECFA、EFSA 及USFDA 等國家或國際組織評估資料優先,若無資料可參考國際上研究文獻,常引用之國家或國際組織說明如下:
1.JECFA:國際食品添加物的安全性評估由JECFA 進行,每年於WHO食品添加物系列(Food Additives Series, FAS)與技術報告(TechnicalReport Series, TRS)公佈安全性評估,各食品添加物JECFA 的評估可在國際化學品安全規劃署(International Programme on Chemical Safety,IPCS)之INCHEM 資料庫(https://inchem.org/#/)中搜尋。
2.EFSA 與SCF:歐盟的食品添加物之風險評估由EFSA 進行,風險評估之科學意見(Scientific Opinion) 結果, 在EFSA 的網頁
(https://www.efsa.europa.eu/en/publications)上公佈。此外在EFSA 設立之前歐盟食品添加物之安全性風險評估由SCF 進行,因此若於EFSA網站中無相關風險評估資料, 也可至SCF 之網站(https://ec.europa.eu/food/horizontal-topics/expert-groups/scientificcommittees/scientific-committee-food-archive_en)搜尋。
3.USFDA:美國的食品添加物風險評估由USFDA 進行,其中屬於GRAS 之食品添加物,在1970 年以前由FDA 進行風險評估,於GRASSubstances (SCOGS) Database 列表中(https://www.fda.gov/food/generally-recognized-safe-gras/grassubstances-
scogs-database)公佈。此風險評估結果在美國技術資訊服務(National Technical Information Service, NTIS)的網站可以付費取得。而1997 年以後的申報GRAS 作為GRAS Notice Inventory 之食品添加物評估內容已公開。
4.FSANZ:在澳洲與紐西蘭的食品添加物的風險評估由FSANZ 進行,風險評估結果是作為Approval Report 在FSANZ 的網頁上公佈。
5.日本食品安全委員會:日本食品安全委員會的食品健康影響評估的結果,作為評估書在食品安全委員會的網頁上公佈。
(九) 依據國人膳食習慣之風險評估
依食安法第18 條第2 項:「前項標準之訂定,必須以可以達到預期效果之最小量為限制,且依據國人膳食習慣為風險評估,同時必須遵守規格標準之規定」,即食品添加物標準之訂定,必須依據國人膳食習慣為風險評估制定可維護國人飲食安全之使用規範。若該項目屬申請項目2.B 得免附。
根據FAO 與WHO 共同所提出之「食品中化學物風險評估原則與方法(Principles and Methods for the Risk Assessment of Chemicals in Food)」所述,風險評估考慮所有可供參考之相關科學數據,並在現有知識基礎上蒐集所有不確定因素,用以制定風險管理措施。
風險評估針對食品中化學物質對健康的可能影響,提供一個標準的審查程序與評估機制,評估食品中化學物質對健康的可能影響(FAO/WHO, 2009)。
依據Codex 所提出之「國際食品法典委員會程序手冊」所述,風險評估可分為四大步驟: 危害鑑定(Hazard Identification) 、危害特徵描述(HazardCharacterization)、暴露評估(Exposure Assessment)及風險特徵描述(RiskCharacterization) (FAO/WHO, 2016)。
1.危害鑑定:
是一種定性的風險評估,主要是針對食品添加物之潛在危害進行調查,由於食品添加物在進入人體後常轉化為其他代謝產物,因此在進行危害鑑定時,除了考慮食品添加物本身外,亦須考慮其衍生代謝產物,並經由危害性資料之收集與解析,瞭解相關之物化特性,以評估其可能對人體造成的健康危害。常用之資料庫如下:
(1) 勞動部職業安全衛生署「危害物質危害數據資料庫」
http://ghs.osha.gov.tw/CHT/intro/search.aspx
(2) 美國毒理學網路(TOXNET)
https://www.nlm.nih.gov/toxnet/index.html
(3) 化學物質摘要服務(Chemical Abstracts Service, CAS)
(4)聯合國糧農組織與世界衛生組織食品添加物聯合專家委員會(Joint FAO/WHO Expert Committee on Food Additives, JECFA)
http://apps.who.int/food-additives-contaminantsjecfa- database/search.aspx
(5)歐洲食品安全局(European Food Safety Authority, EFSA)
(6) 美國毒性物質與疾病登記署(Agency for Toxic Substances and Disease Registry, ATSDR)
2.危害特徵描述:
化學物質對人體的毒理參數可分為非致癌毒理試驗與致癌毒理試驗。非致癌毒理試驗其劑量效應關係為非線性關係,而非線性劑量效應評估通常用在危害性化學物質對於健康效應具有閾值的情況。
毒理學試驗大致可分為利用來自實驗動物或人體的器官、細胞或組織進行培養的體外試驗與利用實驗動物或人體進行的體內試驗。
上述試驗有許多研究目的,包括發現潛在的健康危害,確定產生健康危害作用所必需的暴露條件與評估劑量反應關係。劑量反應評估可提供定量或定性風險評估的分析或建立健康準則含量(HBGV)(FAO/WHO, 2009),例如:ADI、MTDI、PMTDI 及RfD,即無明顯健康風險的人體暴露量。常用之資料庫如下:
(1) 聯合國糧農組織與世界衛生組織食品添加物聯合專家委員會 (Joint FAO/WHO Expert Committee on Food Additives, JECFA)
http://apps.who.int/food-additives-contaminants-jecfadatabase/ search.aspxbase/search.aspx
(2)歐洲食品安全局(European Food Safety Authority, EFSA)
(3) 美國環保署整合性風險資料系統(Integrated Risk Information System, IRIS)
http://www.epa.gov/irisv/iris
毒理參數說明:常用的毒理參數有ADI、MTDI 及RfD 等,而其所根據的毒理資料庫各有不同,當中為了得到毒理參數所建立的毒理試驗,更可為風險評估過程增加可信賴度,以下為常用毒理參數之說明。
(1) 起始點(POD)介紹:起始點為制定毒理參數(例如:ADI、RfD 等)的依據,通常使用動物實驗的毒性研究建立,NOAEL 與LOAEL常作為劑量反應評估之POD,但是如果數據足以進行劑量反應模擬,則可以使用BMD 與BMDL 作為POD,引用毒理參數時應提供推導POD 的數據。如果在最高實驗劑量下未觀察到任何影響,實驗的最高劑量為NOAEL,即為該研究之POD,可表示為:NOAEL 為x mg/kg bw-day 或每日x mg/kg bw,即實驗的最高劑量。當在所有劑量下都觀察到反應時,則無法確定NOAEL,在這種情況下,此研究的POD 為LOAEL,可表示為:無法確定NOAEL,因為在所有劑量下皆觀察到反應,LOAEL為x mg/kg bw-day 或每日x mg/kg bw,即實驗的最低劑量。
(2) ADI:為常用的風險管理決策工具,JECFA 將其定義為終生經食物或飲用水攝入某一化學物質不會對攝食者健康造成明顯危害的量,ADI 值是根據評估時所有已知的資訊推導而出,以mg/kg 表示,適用於食品添加物、食品中的農藥殘留與動物用藥殘留(FAO/WHO, 2009) , 美國環保署(US EnvironmentalProtection Agency, USEPA)則定義為人體於可以長時間暴露某一化學物質而沒有受到健康危害的量(IRIS, 1993)。
(3) PMTDI:JECFA 對於一些可在體內蓄積一段時間的污染物建議使用PMTDI。PMTDI 為JECFA 制定的參考值,用來表示具累積毒性污染物的安全攝取量,由於污染物天然存在於食品與飲用水中,故此值代表人體的允許暴露量。對於既是必須營養素又是食物成分的微量元素則以一個範圍來表示,下限代表個體的必須攝取量,上限則為PMTDI。因為通常缺乏人體低劑量暴露的結果,因此耐受攝取量一般為暫訂的數據,新的數據可能會改變這個暫定的耐受攝取量(FAO/WHO, 2009),其中ADI 與PMTDI 均可視為HBGV (FAO/WHO, 2009)。
(4) RfD:估計一般人群與敏感族群終生暴露某物質且可能沒有明顯健康危害作用之每日暴露量,需透過NOAEL 或LOAEL、能夠反映不同類型資料的UF 與基於專業化學數據所訂定的修飾係數(Modifying Factor, MF)等參數計算而得(IRIS, 1992; WHO IPCS, 1996)。
(5) ARfD:對於大多數食品添加物,在與人體接觸相關的劑量下不會產生急性毒性。因此沒有建立ARfD 的必要,也不需要進行急性膳食暴露評估。但在某些情況下可能與急性不耐受反應相關,例如:糖醇甜味劑引起的腹瀉。
毒理參數引用說明:常用的毒理參數有ADI、MTDI 及RfD 等,而其所根據的毒理資料庫各有不同,當中為了得到毒理參數所建立的毒理試驗,更可為風險評估過程增加可信賴度,為方便業者蒐集毒理參數,本指引以苯甲酸、磷酸鹽類及亞硝酸鹽為例,說明進行毒理參數之引用步驟,舉例說明如下:
(1) 苯甲酸:以苯甲酸鈉為例,需至JECFA 網站,首先須輸入化學名稱(Chemical Name)或CAS 編號(CASRN)進行搜尋,即可根據搜尋結果內之ADI 值填寫,填寫後點選毒理專刊(Tox Monograph),並輸入化學名稱(Chemical Name)進行搜尋,即可根據搜尋結果內之評估(Evaluation),再詳細閱讀與引用。
(2) 磷酸鹽類:以多磷酸鈉為例,需至JECFA 網站,首先須輸入化學名稱(Chemical Name)或CAS 編號(CASRN)進行搜尋,即可根據搜尋結果內之MTDI 值填寫,填寫後點選毒理專刊(Tox Monograph),並輸入化學名稱(Chemical Name)進行搜尋,即可根據搜尋結果內之評估(Evaluation),再詳細閱讀與引用。
(3) 亞硝酸鹽:以亞硝酸鈉為例,需至JECFA 網站,首先須輸入化學名稱(Chemical Name)或CAS 編號(CASRN)進行搜尋,即可根據搜尋結果內之ADI 值填寫,填寫後點選毒理專刊(Tox Monograph),並輸入化學名稱(Chemical Name)進行搜尋,即可根據搜尋結果內之評估(Evaluation),再詳細閱讀與引用。另也可至IRIS 網站進行毒理參數搜尋與引用,首先須輸入化學名稱(Chemical Name)或CAS 編號(CASRN)進行搜尋,即可根據搜尋結果內之RfD 值填寫,並下載其所提供之佐證文件(Summary Document),根據文件內所提及之內容再詳細閱讀與引用。
3.暴露評估:
食品添加物經由食品攝入與其他途徑進入人體後,所進行之人體暴露量定量評估。經計算食品添加物之EDI 而得,需要之濃度、攝食量及體重等暴露參數說明如下:
(1) 暴露族群:風險評估結果會因不同暴露族群有不同結果或需特別關注,本指引參考國家攝食資料庫(https://tnfcds.nhri.edu.tw/ )之建議,將暴露族群定為8 個年齡層,分別為0-3 歲、3-6歲、6-12 歲、12-16 歲、16-18 歲、18-65 歲、65 歲以上及19-49 歲育齡婦女,除19-49 歲育齡婦女,其餘各年齡層依性別再分為男性、女性及全體,一共為22 個暴露族群。
(2) 食品中食品添加物濃度:若業者申請增列食品添加物新品項,可引用國際上食品添加物之實際使用濃度文獻、總膳食研究資料、後市場調查資料、自行檢驗資料、國際間食品添加物法規允許之限量標準等;若業者申請修正現行使用範圍及用量標準,則需同時代入欲修正之食品添加物申請修正濃度與現行食品添加物規限量標準代入計算。
(3) 食品攝食量:可參考國家攝食資料庫之攝食量資料,將食物分類為17 大項、67 小類、199 細項及131 食物品項,可依據業者欲申請增修訂之食物分類、將上述22 個暴露族群,分別依全體族群(Whole Group)與僅攝食者族群(Consumers Only)攝食量計算,「全體族群」為考慮全體族群食用該食品之攝食量數據,「僅攝食者族群」為僅列入有食用該食品之受訪者攝食量數據。
(4) 族群平均體重:可參考國家攝食資料庫之不同族群體重。
(5) EDI:將目標食品的每日攝食量乘以所對應食品中食品添加物添加量計算。若業者申請修正現行使用範圍及用量標準則須同時代入欲修正之使用限量與現行食品添加物法規限量標準。
可使用國家攝食資料庫或其他相關來源的食物攝食量數據進行適當計。食品添加物的安全性應通過將每日攝取劑量與通過毒性試驗確定的每日可接受攝取劑量進行比較。當同時使用相似的食品添加物時,此食品添加物的安全性也應一同評估,食品中食品添加物之濃度可填寫實際使用濃度或法規限量標準。暴露族群經由攝食途徑暴露食品添加物之EDI 可依下列公式計算:
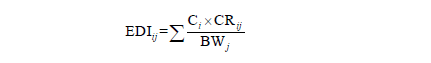
式中,EDIij:暴露族群j 暴露食品i 中食品添加物之估計每日攝取劑量(mg/kg-day),Ci:食品i 中食品添加物之濃度(mg/kg),CRij:暴露族群j 攝食食品i 之每日攝食量(g/day),BWj:不同暴露族群之平均體重(kg)。
4.風險特徵描述:
整合危害鑑定、危害特徵描述及暴露評估之結果,將風險評估結果進行定性與定量做描述。本計畫以待評估食品添加物之EDI 與ADI 數值比較,評估食品添加物在膳食中之風險,以%ADI 表示,風險計算結果需探討其不確定性,暴露族群經由攝食途徑攝入食品添加物之%ADI 可依下列公式計算之:
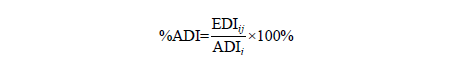
式中,%ADI:暴露食品添加物i 之估計每日攝取劑量佔每日可接受劑量之百分比。ADI:每日可接受劑量(mg/kg-day)。若計算出之風險小於100 %ADI 則為可接受之風險,若風險計算結果大於100 %ADI,則該食品添加物於欲申請之使用範圍與使用限量下具有潛在風險,將不予通過。
若於JECFA 與EFSA 等國際組織無法蒐集到ADI 或僅有暫定ADI(Temporary ADI),可能為以下情況:
(1) 未訂定ADI (Not Specified):若JECFA 認為以數字形式設置ADI 不合適,當食品添加物的估計膳食暴露預期遠低於通常分配的數值時,就會出現這種情況,在此情況下,JECFA 將未訂定ADI。
(2) 暫定ADI 與未訂定ADI (Temporary ADI and ADI Not Specified):若JECFA 評估食品添加物的現有數據存在限制,或者之前訂定的食品添加物ADI 之安全性受到新數據質疑,在制訂與評估進一步安全數據所需要相對較短的時間內使用該物質是安全的,但不確定其在整個生命週期內的使用是否安全時,通常會建立暫定ADI,等待適當的數據提交,以根據JECFA確定的時間表來解決安全問題。
在建立暫定ADI 時,委員會使用較高的不確定性因子,通常增加2 倍,並對額外所需要的生化或毒理學數據所建立ADI 會有明確說明,而且會在暫定期限屆滿前對這些新數據進行審查。在許多情況下,需要進行長期研究,無法按時間表進行,這表示JECFA 不得不將暫定ADI 延長一段時間,若缺乏數據,JECFA 會撤銷暫定ADI,作為安全防範措施。
另應進行不確定性分析(Uncertainty Analysis),不確定性分析代表對事件或訊息的無法肯定、缺乏正確、或完整的知識(Lack of
Knowledge)來佐證,或是錯誤的統計造成的偏差(Bias),故不確定性分析是指風險評估者在瞭解所使用資料與評估模型的局限性,描述暴露與風險評估中的不確定性。
進行不確定性分析時,需明確描述風險評估中不確定性的主要來源,並進行半定量評估,例如:高估與/或低估,判斷風險是否符合保守性。可參考EFSA 於FAIM 之評估方式,依不確定性之來源可分為三類:情境、參數及模式,根據EFSA (2012)對膳食暴露不確定性之指導建議可參考以下範例。
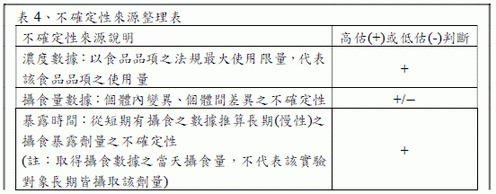
(十) 在人類上之研究與發現
若有人類上之研究與發現,例如:志願者研究(Volunteer Studies)、流行病學研究(Epidemiology Studies)、職業暴露人員監測(Health EffectStudies Relating to Occupational Exposure)、急性中毒案例研究及臨床試驗數據(Clinical Experience),得以引用。此外在懷疑可能有致過敏性的情況下,無法由動物試驗結果外推人類,因此在人類上之研究與發現也需重視。
若無適當在人類上之研究與發現數據,得免附,並於後續「無法檢具上述資料之說明」解釋無法取得相關資料之原因。屬於申請項目2.B 修正現行規格標準者得免附。
(十一) 其他必要性資料
可供其他佐證之安全性與功能等資料。
(十二) 無法檢具上述資料之說明
若業者無法取得或確認該食品添加物尚無上述相關研究資料,得陳述無法檢具上述各項資料之項目,並簡潔說明無法取得之原因。
四、參考文獻
參考文獻說明請參閱本指引貳、二、(四)內容。
肆、參考文獻
European Union (EU), 2020, Practical Guidance for Applicants on the Submission of Applications on Food Additives, Food Enzymes and Food Flavourings.
FAO/WHO, 2009. Principles and Methods for the Risk Assessment of Chemicals in Food. Environmental Health Criteria 240.
https://www.who.int/publications/i/item/9789241572408
Integrated Risk Information System (IRIS), 1993. Reference Dose (RfD): Description and Use in Health Risk Assessments. Washington, DC, US Environmental Protection Agency.
https://www.epa.gov/iris/reference-dose-rfd-description-and-usehealth-risk-assessments
Integrated Risk Information System (USEPA IRIS), 1992. Glossary of Risk Assessment-Related Terms. Washington, DC, US Environmental Protection Agency.
https://www.epa.gov/iris/iris-glossary
Joint FAO/WHO Expert Committee on Food Additives (JECFA), 2016, Guidance Document for WHO Monographers and Reviewers Evaluating Food Additives.
https://www.who.int/publications-detail-redirect/9789241511155
U.S. Food and Drug Administration (USFDA), 2009, Guidance for Industry: Recommendations for Submission of Chemical and Technological Data for Direct Food Additive Petitions.
日本厚生勞動省醫藥食品局,1996,食品添加物の指定及び使用基準改正要請資料作成に関する手引。
食品藥物管理署110 年12 月公布之「食品中化學性物質風險評估參考手冊」
http://www.fda.gov.tw/TC/site.aspx?sid=12149&r=798796427
伍、附件
附件一、「食品添加物使用範圍及限量暨規格標準」增修訂申請表


附件二、Application Form of Additions and Amendments to the Scope of Use, Limits and Specifications of Food Additives



資料來源:衛生福利部食品藥物管理署
如您有食品查驗登記、成分包裝諮詢的需求,綠霖生技顧問為您提供最專業的諮詢服務,您可經由下方的按鈕與綠霖聯繫